
By: Mady Docteur, Paige Hoen and Hailey Ramthun
11 October 2024
This semester, we have continued to research our three genes: ABHD2, DBP and PALD1 and how they are expressed in green anole lizards (Anolis carolinensis). The goal of this research is to see gene expression differences between the breeding and non-breeding seasons of the lizards. Last semester, we found a successful primer set for both ABHD2 and DBP. We also found three successful primer sets for the gene PALD1.
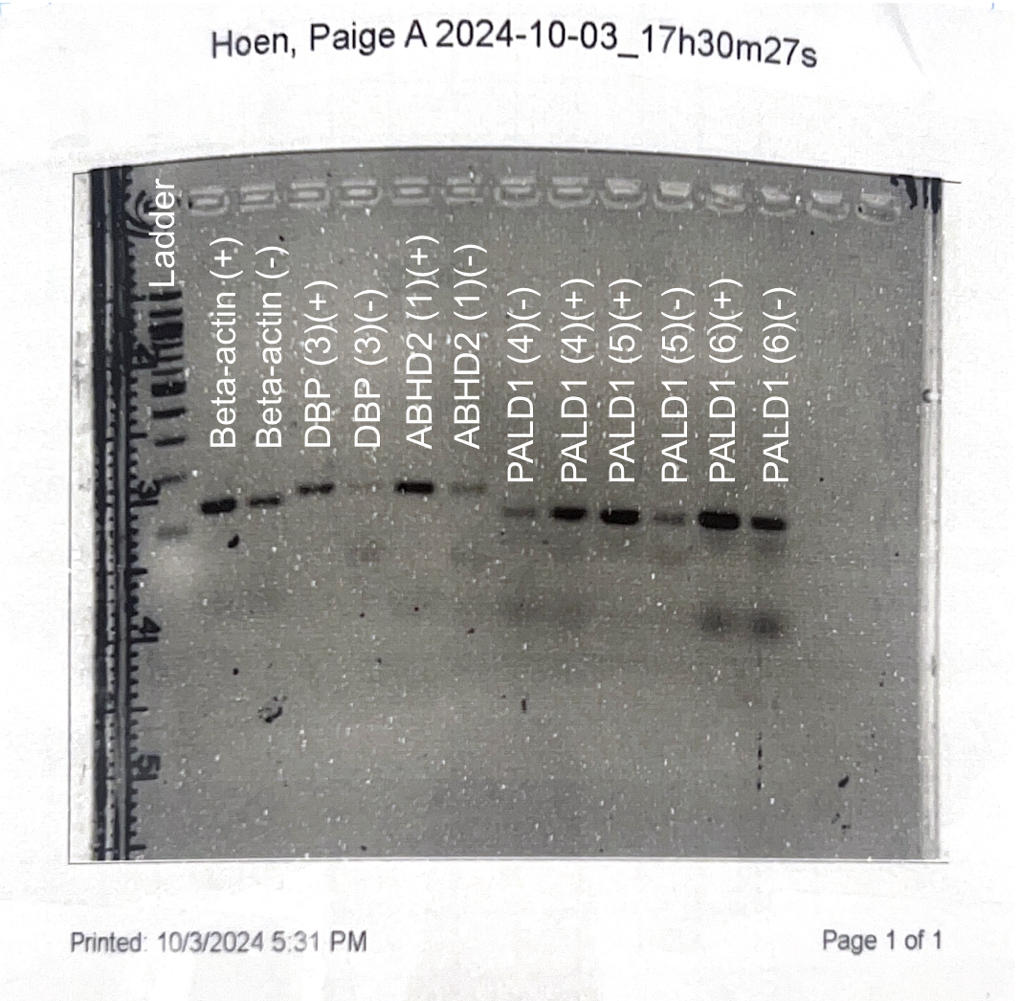
In the past two weeks, we have been ensuring all these primer sets still work through PCR and gel electrophoresis. After performing gel electrophoresis, we image each gel to check that the correct band size is amplified for each primer set. In our last gel image, all our primer sets amplified the correct band size; however, a lot of primer dimers were present. Although primer dimers are not ideal to have in a gel, we decided to move on to qPCR.
Last week, we set up a qPCR for the first time. The difference between qPCR and PCR is that qPCR allows us to monitor a PCR reaction in real time and receive more detailed results. When setting up a qPCR, we create multiple serial dilutions of a master mix to create a standard curve. Then, we load each dilution into a 96-well plate.
After loading the plate with our samples, we perform the qPCR software protocol. We start by setting up our experiment on a computer to collect data. In this data, an important value we look for is our PCR efficiency for each primer set. Unfortunately, our qPCR showed PCR efficiency values that were much higher than they should be. This could be due to primer dimers or pipetting error when setting up the qPCR.

After running our qPCR plate, we performed gel electrophoresis and loaded each of our samples into a gel. After imaging the gel, it showed numerous primer dimers. This could have been due to inaccurate pipetting or adding contaminated cDNA when preparing the qPCR samples.
Our next step will be setting up another qPCR. We will use different cDNA to try to limit primer dimers and focus more on pipetting carefully. We are hoping to end with a lower PCR efficiency value. If the results of our next qPCR are not what we are looking for, we will begin troubleshooting. The past couple of weeks have taught us that research does not always go smoothly. Results do not always come back how we want them to, but this allows us to practice perseverance despite challenges and learn troubleshooting skills.